An estimated seven to ten million people worldwide are living with Parkinson’s disease (PD), which is an incurable and progressive disease of the nervous system affecting movement and cognitive function. More than half of PD patients develop progressive disease showing signs of dementia similar to Alzheimer’s disease. A research team at University of Copenhagen, Denmark, has discovered that non-inheritable PD may be caused by functional changes in the immune regulating gene Interferon-beta (IFNβ). Treatment with IFNβ-gene therapy successfully prevented neuronal death and disease effects in an experimental model of PD. The results have just been published in prestigious scientific journal Cell.
Protein regulates waste management in nerve cells
The human brain consists of approximately 100 billion neurons, which coordinate activities in all parts of the body. At Biotech Research and Innovation Centre (BRIC), University of Copenhagen, the group of Professor Shohreh Issazadeh-Navikas has discovered that the immune gene IFNβ plays a vital role in keeping neurons healthy.
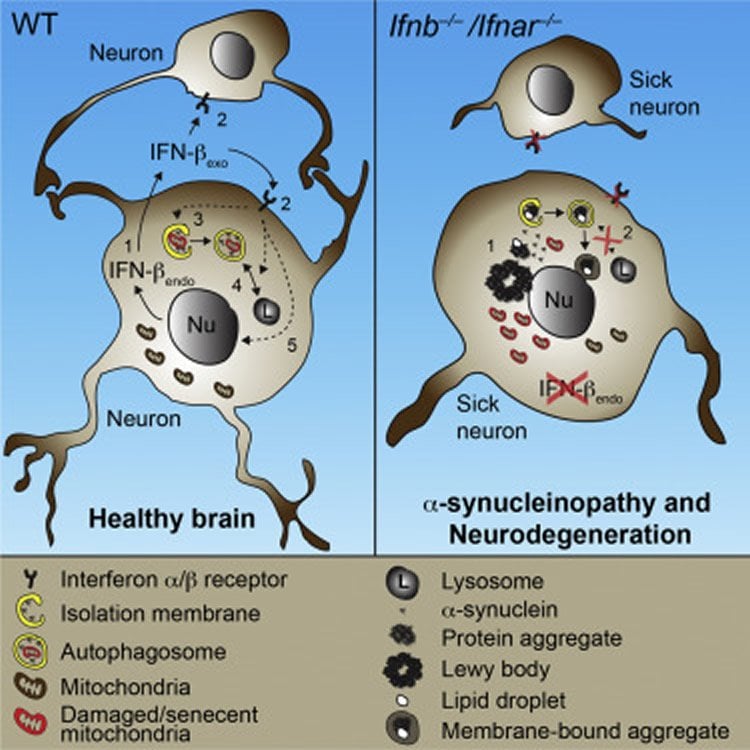
“We found that IFNβ is essential for neurons ability to recycle waste proteins. Without this, the waste proteins accumulate in disease-associated structures called Lewy bodies and with time the neurons die”, explains assistant professor Patrick Ejlerskov, the first author of this report.
The research team found that mice missing IFNβ developed Lewy bodies in parts of the brain, which control body movement and restoration of memory, and as a result they developed disease and clinical signs similar to patients with PD and dementia with Lewy bodies (DLB).
New understanding of non-familial Parkinson’s disease
While hereditary gene mutations have long been known to play a role in familial PD, the study from BRIC offers one of the first models for so-called non-familial PD, which comprises the majority (90-95%) of patients suffering from PD. According to professor Shohreh Issazadeh-Navikas the new knowledge opens new therapeutic possibilities.
“This is one of the first genes found to cause pathology and clinical features of non-familial PD and DLB, through accumulation of disease-causing proteins. It is independent of gene mutations known from familial PD and when we introduced IFNβ-gene therapy, we could prevent neuronal death and disease development. Our hope is that this knowledge will enable development of more effective treatment of PD”, says professor Shohreh Issazadeh-Navikas.
Current treatments are effective at improving the early motor symptoms of the disease, however, as the disease progress, the treatment effect is lost. The next step for the research team will be to gain a better understanding of the molecular mechanisms by which IFN? protects neurons and thereby prevents movement disorders and dementia.
Funding: The Danish Council For Independent Research, Alzheimer-forskningsfonden, Danish Multiple Sclerosis Society, Danish CancerSociety, and Lundbeck Foundation funded this research.
Source: Patrick Ejlerskov – University of Copenhagen Biotech Research and Innovation Centre
Image Credit: The image is credited to Issazadeh-Navikas et al./Cell 2015
Original Research: Full open access research for “Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia” by Patrick Ejlerskov, Jeanette Göransdotter Hultberg, JunYang Wang, Robert Carlsson, Malene Ambjørn, Martin Kuss, Yawei Liu, Giovanna Porcu, Kateryna Kolkova, Carsten Friis Rundsten, Karsten Ruscher, Bente Pakkenberg, Tobias Goldmann, Desiree Loreth, Marco Prinz, David C. Rubinsztein, and Shohreh Issazadeh-Navikas in Cell. Published online October 8 2015 doi:10.1016/j.cell.2015.08.069
Abstract
Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia
Highlights
•Lack of neuronal IFN-β-IFNAR signaling causes brain Lewy body accumulation
•IFN-β deficiency causes late-stage autophagy block and thereby α-synuclein aggregation
•IFN-β promotes neuronal autophagy and α-synuclein clearance
•Ifnb gene therapy prevents dopaminergic neuron loss in a familial PD model
Summary
Neurodegenerative diseases have been linked to inflammation, but whether altered immunomodulation plays a causative role in neurodegeneration is not clear. We show that lack of cytokine interferon-β (IFN-β) signaling causes spontaneous neurodegeneration in the absence of neurodegenerative disease-causing mutant proteins. Mice lacking Ifnb function exhibited motor and cognitive learning impairments with accompanying α-synuclein-containing Lewy bodies in the brain, as well as a reduction in dopaminergic neurons and defective dopamine signaling in the nigrostriatal region. Lack of IFN-β signaling caused defects in neuronal autophagy prior to α-synucleinopathy, which was associated with accumulation of senescent mitochondria. Recombinant IFN-β promoted neurite growth and branching, autophagy flux, and α-synuclein degradation in neurons. In addition, lentiviral IFN-β overexpression prevented dopaminergic neuron loss in a familial Parkinson’s disease model. These results indicate a protective role for IFN-β in neuronal homeostasis and validate Ifnb mutant mice as a model for sporadic Lewy body and Parkinson’s disease dementia.
“Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia” by Patrick Ejlerskov, Jeanette Göransdotter Hultberg, JunYang Wang, Robert Carlsson, Malene Ambjørn, Martin Kuss, Yawei Liu, Giovanna Porcu, Kateryna Kolkova, Carsten Friis Rundsten, Karsten Ruscher, Bente Pakkenberg, Tobias Goldmann, Desiree Loreth, Marco Prinz, David C. Rubinsztein, and Shohreh Issazadeh-Navikas in Cell. Published online October 8 2015 doi:10.1016/j.cell.2015.08.069