

Summary: A new Nature study reports blocking ApoE4 may help prevent inflammation and cell death that contribute to Alzheimer’s disease.
Source: WUSTL.
Nearly a quarter century ago, a genetic variant known as ApoE4 was identified as a major risk factor for Alzheimer’s disease — one that increases a person’s chances of developing the neurodegenerative disease by up to 12 times.
However, it was never clear why the ApoE4 variant was so hazardous. When the ApoE4 protein is present, clumps of the protein amyloid beta accumulate in the brain. But such clumps alone do not kill brain cells or lead to characteristic Alzheimer’s symptoms such as memory loss and confusion.
Now, a study led by researchers at Washington University School of Medicine in St. Louis shows that the presence of ApoE4 exacerbates the brain damage caused by toxic tangles of a different Alzheimer’s-associated protein: tau. In the absence of ApoE, tau tangles did very little harm to brain cells.
The findings suggest that targeting ApoE could help prevent or treat the brain damage present in Alzheimer’s disease, for which there are currently no effective therapies.
“Once tau accumulates, the brain degenerates,” said senior author David Holtzman, MD, the Andrew B. and Gretchen P. Jones Professor and head of the Department of Neurology. “What we found was that when ApoE is there, it amplifies the toxic function of tau, which means that if we can reduce ApoE levels we may be able to stop the disease process.”
The study is published Sept. 20 in the journal Nature.
Alzheimer’s, which affects one in 10 people over age 65, is the most common example of a family of diseases called tauopathies. The group also includes chronic traumatic encephalopathy, which plagues professional boxers and football players, and several other neurodegenerative diseases.
To find out what effect ApoE variants have on tauopathies, Holtzman and graduate student Yang Shi and their colleagues turned to genetically modified mice that carry a mutant form of human tau prone to forming toxic tangles.
They utilized mice that lacked their own version of the mouse ApoE gene or replaced it with one of the three variants of the human ApoE gene: ApoE2, ApoE3 or ApoE4. Compared with the majority of people who have the more common ApoE3 variant, people with ApoE4 are at elevated risk of developing Alzheimer’s, and those with ApoE2 are protected from the disease.
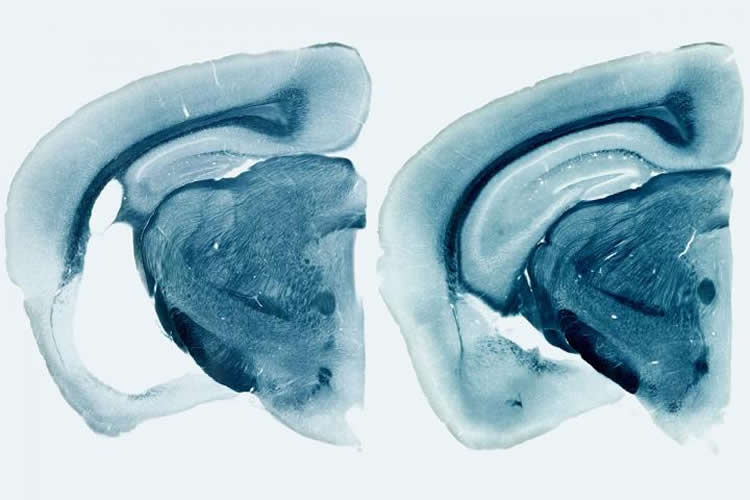
By the time the mice were 9 months old, the ones carrying human ApoE variants had widespread brain damage. The hippocampus and entorhinal cortex, important for memory, were shrunken, and the fluid-filled space of the brain had enlarged where the dead cells had been. ApoE4 mice exhibited the most severe neurodegeneration, and ApoE2 the least. The mice that lacked ApoE entirely showed virtually no brain damage.
Further, the immune cells in the brains of mice with ApoE4 turned on a set of genes related to activation and inflammation much more strongly than those from ApoE3 mice. Immune cells from mice lacking ApoE were barely activated.
“ApoE4 seems to be causing more damage than the other variants because it is instigating a much higher inflammatory response, and it is likely the inflammation that is causing injury,” Holtzman said. “But all forms of ApoE – even ApoE2 – are harmful to some extent when tau is aggregating and accumulating. The best thing seems to be in this setting to have no ApoE at all in the brain.”
To find out whether ApoE in people similarly exacerbates neuronal damage triggered by tau, the researchers collaborated with Bill Seeley, MD, from the University of California, San Francisco. Seeley identified autopsy samples from 79 people who had died from tauopathies other than Alzheimer’s disease in the past 10 years. The researchers examined each brain for signs of injury and noted the deceased’s ApoE variants. They found that, at the time of death, people with ApoE4 had more damage than those that lacked ApoE4.
ApoE transports cholesterol around the body via the bloodstream. A few, rare individuals lack a functional ApoE gene. Such people have very high cholesterol levels and, if untreated, die young of cardiovascular disease. The lack of ApoE in their brains, however, creates no obvious problems.
“There are people walking around who have no ApoE and they’re fine cognitively,” Holtzman said. “It doesn’t appear to be required for normal brain function.”
These findings suggest that decreasing ApoE specifically in the brain could slow or block neurodegeneration, even in people who already have accumulated tau tangles. Most investigational therapies for Alzheimer’s disease have focused on amyloid beta or tau, and none has been successful yet in changing the trajectory of the disease. Targeting ApoE has not yet been tried, according to Holtzman.
“Assuming that our findings are replicated by others, I think that reducing ApoE in the brain in people who are in the earliest stages of disease could prevent further neurodegeneration,” Holtzman said.
Funding: Funding provided by National Institutes of Health, JPB Foundation, Cure Alzheimer’s Fund, AstraZeneca, Consortium for Frontotemporal Dementia Research, Tau Consortium, National Multiple Sclerosis Society, Nancy Davis Foundation Award, and others.
Source: Judy Martin Finch – WUSTL
Image Source: NeuroscienceNews.com image is credited to Yang Shi.
Original Research: Abstract for “ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy” by Yang Shi, Kaoru Yamada, Shane Antony Liddelow, Scott T. Smith, Lingzhi Zhao, Wenjie Luo, Richard M. Tsai, Salvatore Spina, Lea T. Grinberg, Julio C. Rojas, Gilbert Gallardo, Kairuo Wang, Joseph Roh, Grace Robinson, Mary Beth Finn, Hong Jiang, Patrick M. Sullivan, Caroline Baufeld, Michael W. Wood, Courtney Sutphen, Lena McCue, Chengjie Xiong, Jorge L. Del-Aguila, John C. Morris, Carlos Cruchaga, Alzheimer’s Disease Neuroimaging Initiative, Anne M. Fagan, Bruce L. Miller, Adam L. Boxer, William W. Seeley, Oleg Butovsky, Ben A. Barres, Steven M. Paul & David M. Holtzman in Nature. Published online September 20 2017 doi:10.1038/nature24016
[cbtabs][cbtab title=”MLA”]WUSTL “Newly Identified Role of ApoE4 Suggests Possible Therapeutic Target for Alzheimer’s.” NeuroscienceNews. NeuroscienceNews, 20 September 2017.
<https://neurosciencenews.com/apoe4-genetics-alzheimers-7529/>.[/cbtab][cbtab title=”APA”]WUSTL (2017, September 20). Newly Identified Role of ApoE4 Suggests Possible Therapeutic Target for Alzheimer’s. NeuroscienceNews. Retrieved September 20, 2017 from https://neurosciencenews.com/apoe4-genetics-alzheimers-7529/[/cbtab][cbtab title=”Chicago”]WUSTL “Newly Identified Role of ApoE4 Suggests Possible Therapeutic Target for Alzheimer’s.” https://neurosciencenews.com/apoe4-genetics-alzheimers-7529/ (accessed September 20, 2017).[/cbtab][/cbtabs]
Abstract
ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy
APOE4 is the strongest genetic risk factor for late-onset Alzheimer disease. ApoE4 increases brain amyloid-β pathology relative to other ApoE isoforms1. However, whether APOE independently influences tau pathology, the other major proteinopathy of Alzheimer disease and other tauopathies, or tau-mediated neurodegeneration, is not clear. By generating P301S tau transgenic mice on either a human ApoE knock-in (KI) or ApoE knockout (KO) background, here we show that P301S/E4 mice have significantly higher tau levels in the brain and a greater extent of somatodendritic tau redistribution by three months of age compared with P301S/E2, P301S/E3, and P301S/EKO mice. By nine months of age, P301S mice with different ApoE genotypes display distinct phosphorylated tau protein (p-tau) staining patterns. P301S/E4 mice develop markedly more brain atrophy and neuroinflammation than P301S/E2 and P301S/E3 mice, whereas P301S/EKO mice are largely protected from these changes. In vitro, E4-expressing microglia exhibit higher innate immune reactivity after lipopolysaccharide treatment. Co-culturing P301S tau-expressing neurons with E4-expressing mixed glia results in a significantly higher level of tumour-necrosis factor-α (TNF-α) secretion and markedly reduced neuronal viability compared with neuron/E2 and neuron/E3 co-cultures. Neurons co-cultured with EKO glia showed the greatest viability with the lowest level of secreted TNF-α. Treatment of P301S neurons with recombinant ApoE (E2, E3, E4) also leads to some neuronal damage and death compared with the absence of ApoE, with ApoE4 exacerbating the effect. In individuals with a sporadic primary tauopathy, the presence of an ε4 allele is associated with more severe regional neurodegeneration. In individuals who are positive for amyloid-β pathology with symptomatic Alzheimer disease who usually have tau pathology, ε4-carriers demonstrate greater rates of disease progression. Our results demonstrate that ApoE affects tau pathogenesis, neuroinflammation, and tau-mediated neurodegeneration independently of amyloid-β pathology. ApoE4 exerts a ‘toxic’ gain of function whereas the absence of ApoE is protective.
“ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy” by Yang Shi, Kaoru Yamada, Shane Antony Liddelow, Scott T. Smith, Lingzhi Zhao, Wenjie Luo, Richard M. Tsai, Salvatore Spina, Lea T. Grinberg, Julio C. Rojas, Gilbert Gallardo, Kairuo Wang, Joseph Roh, Grace Robinson, Mary Beth Finn, Hong Jiang, Patrick M. Sullivan, Caroline Baufeld, Michael W. Wood, Courtney Sutphen, Lena McCue, Chengjie Xiong, Jorge L. Del-Aguila, John C. Morris, Carlos Cruchaga, Alzheimer’s Disease Neuroimaging Initiative, Anne M. Fagan, Bruce L. Miller, Adam L. Boxer, William W. Seeley, Oleg Butovsky, Ben A. Barres, Steven M. Paul & David M. Holtzman in Nature. Published online September 20 2017 doi:10.1038/nature24016




