

Summary: Study reports an accumulation of manganese in cells may disrupt protein transportation, resulting in Parkinsonian syndrome.
Source: CNRS.
Using X-ray fluorescence at synchrotrons DESY and ESRF, researchers in the Centre d’Etudes Nucléaires de Bordeaux Gradignan (CNRS/Université de Bordeaux) have demonstrated the consequences of a mutation responsible for a hereditary parkinsonian syndrome: accumulated manganese in the cells appears to disturb protein transport. This work, carried out with colleagues at the University of Texas at Austin (USA), was published in the print issue of ACS Chemical Neuroscience on January 16, 2018.
Parkinsonian syndrome is a set of diseases with symptoms similar to Parkinson’s disease. Some are caused by high quantities of manganese, a metal essential to the body at trace levels. This is especially so for a hereditary form of the disease caused by a genetic mutation responsible for a toxic accumulation of manganese in cells.
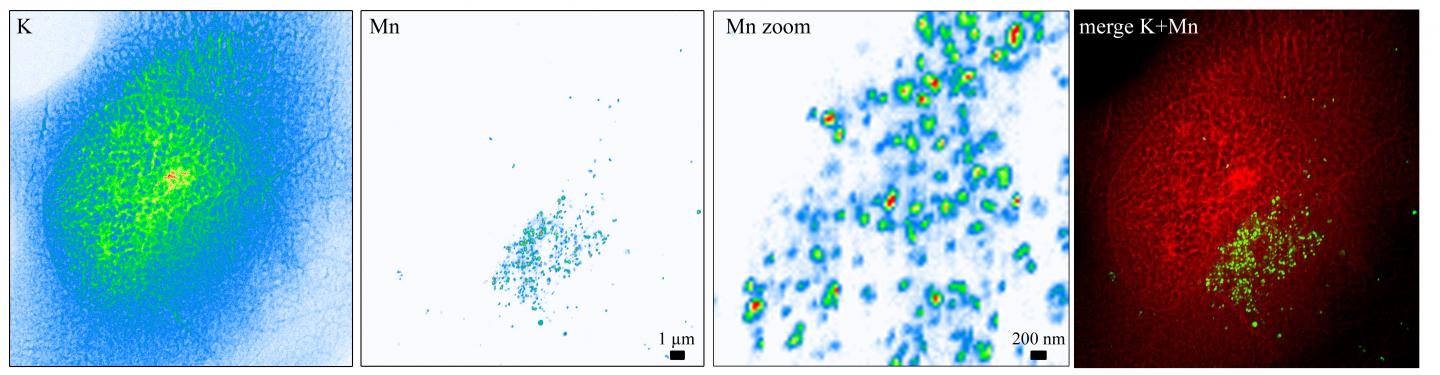
The team of researchers has shown a key mechanism for the disease caused by this mutation. At the DESY synchrotron (Hamburg, Germany), they have been able to locate manganese inside individual cells, (1) using the fluorescent signature it produces under an X-ray beam. Manganese concentrates essentially in the Golgi apparatus, a cellular compartment which acts as a dispatch center for proteins. The proteins receive a label and are accordingly packaged within vesicles to other compartments, or to the outside of the cell. It is in these vesicles–barely 50 nm in diameter–that manganese accumulates, as the researchers have demonstrated by repeating their experiments in the ESRF synchrotron (The European synchrotron, Grenoble), with even higher sensitivity and spatial resolution. This is the only place in the world where the equipment’s spatial resolution and sensitivity were sufficient to detect the minute amounts of manganese in the vesicles.
The researchers think that this manganese accumulation disturbs protein export towards the outside of the cell, altering nerve cell function and leading to parkinsonian symptoms. This must still be confirmed by reproducing these experiments with neurons from animal models for this disease, which are being developed.
Funding: CNRS-IN2P3, NIH/National Institute of Environmental Health Sciences funded this study.
Source: Veronique Etienne – CNRS
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to Asuncion Carmona / CENBG / CNRS.
Original Research: Abstract for “SLC30A10 Mutation Involved in Parkinsonism Results in Manganese Accumulation within Nanovesicles of the Golgi Apparatus” by Asuncion Carmona, Charles E. Zogzas, Stéphane Roudeau, Francesco Porcaro, Jan Garrevoet, Kathryn M. Spiers, Murielle Salomé, Peter Cloetens, Somshuvra Mukhopadhyay, and Richard Ortega in ACS Chemical Neuroscience. Published October 18 2018.
doi:10.1021/acschemneuro.8b00451
[cbtabs][cbtab title=”MLA”]CNRS”How Manganese Produces a Parkinsonian Syndrome.” NeuroscienceNews. NeuroscienceNews, 16 January 2019.
<https://neurosciencenews.com/manganese-parkinsons-10560/>.[/cbtab][cbtab title=”APA”]CNRS(2019, January 16). How Manganese Produces a Parkinsonian Syndrome. NeuroscienceNews. Retrieved January 16, 2019 from https://neurosciencenews.com/manganese-parkinsons-10560/[/cbtab][cbtab title=”Chicago”]CNRS”How Manganese Produces a Parkinsonian Syndrome.” https://neurosciencenews.com/manganese-parkinsons-10560/ (accessed January 16, 2019).[/cbtab][/cbtabs]
Abstract
SLC30A10 Mutation Involved in Parkinsonism Results in Manganese Accumulation within Nanovesicles of the Golgi Apparatus
Manganese (Mn) is an essential metal that can be neurotoxic when elevated exposition occurs leading to parkinsonian-like syndromes. Mutations in the Slc30a10 gene have been identified in new forms of familial parkinsonism. SLC30A10 is a cell surface protein involved in the efflux of Mn and protects the cell against Mn toxicity. Disease-causing mutations block the efflux activity of SLC30A10, resulting in Mn accumulation. Determining the intracellular localization of Mn when disease-causing SLC30A10 mutants are expressed is essential to elucidate the mechanisms of Mn neurotoxicity. Here, using organelle fluorescence microscopy and synchrotron X-ray fluorescence (SXRF) imaging, we found that Mn accumulates in the Golgi apparatus of human cells transfected with the disease-causing SLC30A10-Δ105–107 mutant under physiological conditions and after exposure to Mn. In cells expressing the wild-type SLC30A10 protein, cellular Mn content was low after all exposure conditions, confirming efficient Mn efflux. In nontransfected cells that do not express endogenous SLC30A10 and in mock transfected cells, Mn was located in the Golgi apparatus, similarly to its distribution in cells expressing the mutant protein, confirming deficient Mn efflux. The newly developed SXRF cryogenic nanoimaging (<50 nm resolution) indicated that Mn was trapped in single vesicles within the Golgi apparatus. Our results confirm the role of SLC30A10 in Mn efflux and the accumulation of Mn in cells expressing the disease-causing SLC30A10-Δ105–107 mutation. Moreover, we identified suborganelle Golgi nanovesicles as the main compartment of Mn accumulation in SLC30A10 mutants, suggesting interactions with the vesicular trafficking machinery as a cause of the disease. [divider]Feel free to share this Neuroscience News.[/divider]




