

PKC alpha is required for pathological consequences of amyloid beta plaques; mutations that enhance its activity found in patients with the disease.
In Alzheimer’s disease, plaques of amyloid beta protein accumulate in the brain, damaging connections between neurons. Now, researchers at University of California San Diego School of Medicine and Harvard Medical School have found that the enzyme Protein Kinase C (PKC) alpha is necessary for amyloid beta to damage neuronal connections. They also identified genetic variations that enhance PKC alpha activity in patients with Alzheimer’s disease.
The study, published May 10 in Science Signaling, may present a new therapeutic target for the disease.
“Until recently, it was thought that PKC helped cells survive, and that too much PKC activity led to cancer. Based on that assumption, many companies tested PKC inhibitors as drugs to treat cancer, but they didn’t work,” said co-senior author Alexandra Newton, PhD, professor of pharmacology at UC San Diego School of Medicine.
“Instead, we recently found that the opposite is true. PKC serves as the brakes to cell growth and survival, so cancer cells benefit when PKC is inactivated. Now, our latest study reveals that too much PKC activity is also bad, driving neurodegeneration. This means that drugs that failed in clinical trials for cancer may provide a new therapeutic opportunity for Alzheimer’s disease.”
The study was a three-way collaboration between experts in PKC (Newton), neuroscience (Roberto Malinow, MD, PhD, Distinguished Professor of Neurosciences and Neurobiology at UC San Diego School of Medicine), and genomics (Rudolph Tanzi, PhD, professor of neurology at Harvard Medical School).
Malinow’s team found that when mice are missing the PKC alpha gene, neurons functioned normally, even when amyloid beta was present. Then, when they restored PKC alpha, amyloid beta once again impaired neuronal function. In other words, amyloid beta doesn’t inhibit brain function unless PKC alpha is active.
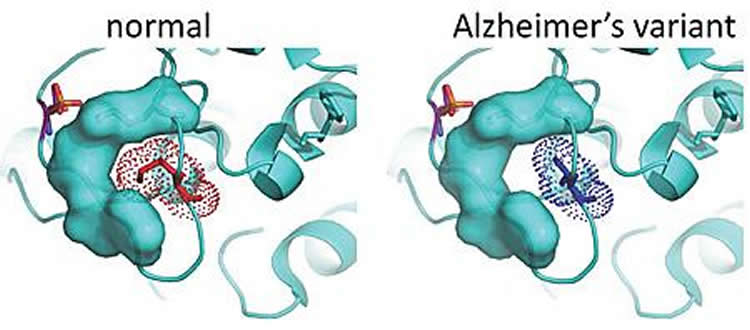
Enter the Tanzi team, which has a database of genetic information for 1,345 people in 410 families with late-onset Alzheimer’s disease. Tanzi and team use this database to look for rare variants — genetic mutations found only in family members with the disease. Here, the team found three variants in one form of the PKC enzyme, PKC alpha that were associated with the disease in five families.
The researchers replicated these three PKC alpha gene variants in laboratory cell lines. In each instance, PKC alpha activity was increased.
While this study surfaced only five families with these rare mutations in the PKC alpha gene, there are many ways to influence PKC alpha’s activity, Newton said. She believes there could be many other inherited genetic variations that indirectly boost or inhibit PKC activity, and therefore also influence a person’s likelihood of developing Alzheimer’s disease.
“Next we want to identify more molecules participating in the pathophysiology,” said Malinow. “The more steps in the mechanism we can understand, the more therapeutic targets we’ll find for Alzheimer’s disease.”
Co-authors of this study also include: Stephanie Alfonso, Julia A. Callender, Corina E. Antal, UC San Diego; Basavaraj Hooli, Kristina Mullin, Harvard Medical School; Mathew A. Sherman, Sylvain E. Lesné, University of Minnesota; and Michael Leitges, University of Oslo.
Funding: This research was funded, in part, by the National Institutes of Health (grants GM43154, MH060009, AG032132, GM007752, DGE1144086) and Cure Alzheimer’s Fund (grant 2015-3999).
Source: Heather Buschman – UCSD
Image Source: The image is credited to UC San Diego Health.
Original Research: Abstract for “Gain-of-function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer’s diseaseg” by Stephanie I. Alfonso, Julia A. Callender, Basavaraj Hooli, Corina E. Antal, Kristina Mullin, Mathew A. Sherman, Sylvain E. Lesné, Michael Leitges, Alexandra C. Newton, Rudolph E. Tanzi, and Roberto Malinow in Science Signaling. Published online May 10 2016 doi:10.1126/scisignal.aaf6209
Abstract
Gain-of-function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive dementia disorder characterized by synaptic degeneration and amyloid-β (Aβ) accumulation in the brain. Through whole-genome sequencing of 1345 individuals from 410 families with late-onset AD (LOAD), we identified three highly penetrant variants in PRKCA, the gene that encodes protein kinase Cα (PKCα), in five of the families. All three variants linked with LOAD displayed increased catalytic activity relative to wild-type PKCα as assessed in live-cell imaging experiments using a genetically encoded PKC activity reporter. Deleting PRKCA in mice or adding PKC antagonists to mouse hippocampal slices infected with a virus expressing the Aβ precursor CT100 revealed that PKCα was required for the reduced synaptic activity caused by Aβ. In PRKCA−/− neurons expressing CT100, introduction of PKCα, but not PKCα lacking a PDZ interaction moiety, rescued synaptic depression, suggesting that a scaffolding interaction bringing PKCα to the synapse is required for its mediation of the effects of Aβ. Thus, enhanced PKCα activity may contribute to AD, possibly by mediating the actions of Aβ on synapses. In contrast, reduced PKCα activity is implicated in cancer. Hence, these findings reinforce the importance of maintaining a careful balance in the activity of this enzyme.
“Gain-of-function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer’s diseaseg” by Stephanie I. Alfonso, Julia A. Callender, Basavaraj Hooli, Corina E. Antal, Kristina Mullin, Mathew A. Sherman, Sylvain E. Lesné, Michael Leitges, Alexandra C. Newton, Rudolph E. Tanzi, and Roberto Malinow in Science Signaling. Published online May 10 2016 doi:10.1126/scisignal.aaf6209




