

A long-term reduction in neuronal activity reduces amyloid plaques associated with Alzheimer’s disease, Yale University researchers have found. The study, using mouse models of Alzheimer’s, found the opposite is also true — triggering an increase in neuronal activity spurs creation of plaques and toxic amyloid beta peptides believed to trigger the disease.
In the accompanying image, amyloid plaques in blue are surrounded by damaged neuronal branches.
Neuroscientists Jaime Grutzendler and Peng Yuan used a new technology called chemogenetics to alternately inhibit or excite neuronal signaling in mice with Alzheimer’s. The goal was to test the theory that hyperactivity in neurons might contribute to the onset of Alzheimer’s. Such an increase in neuronal activity might trigger the production of excess amounts of amyloid beta peptides, which in turn may be toxic to brain cells and cause the symptoms of dementia, Grutzendler said.
On the other hand, suppressing this hyperactivity could be a novel way of halting the progression of the disease, he noted.
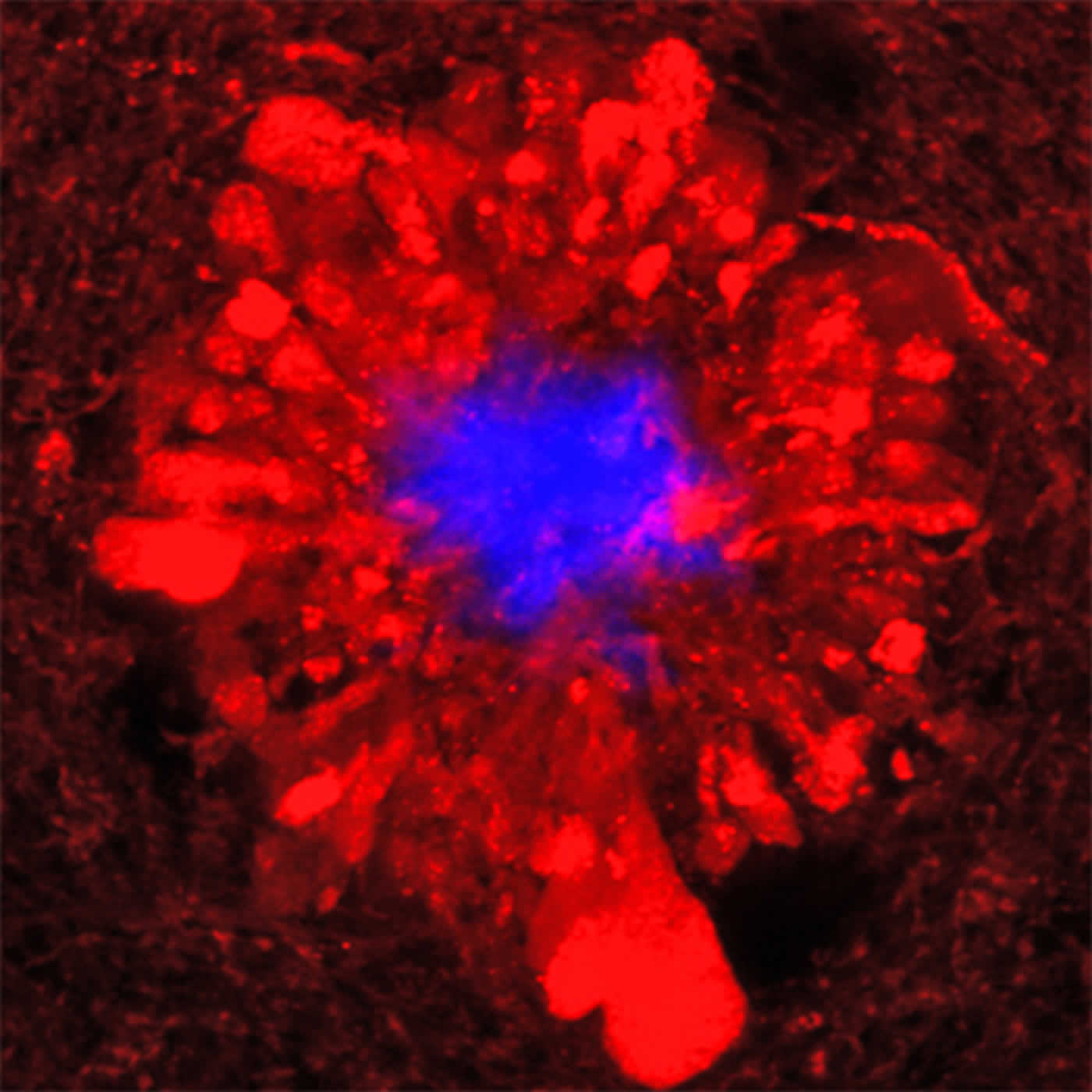
Source: Bill Hathaway – Yale
Image Credit: The image is adapted from the Yale press release
Original Research: Abstract for “Attenuation of β-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity” by Peng Yuan (袁鹏) and Jaime Grutzendler in Journal of Neuroscience. Published online January 13 2016 doi:10.1523/JNEUROSCI.2531-15.2016
Abstract
Attenuation of β-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity
Aberrant neural hyperactivity has been observed in early stages of Alzheimer’s disease (AD) and may be a driving force in the progression of amyloid pathology. Evidence for this includes the findings that neural activity may modulate β-amyloid (Aβ) peptide secretion and experimental stimulation of neural activity can increase amyloid deposition. However, whether long-term attenuation of neural activity prevents the buildup of amyloid plaques and associated neural pathologies remains unknown. Using viral-mediated delivery of designer receptors exclusively activated by designer drugs (DREADDs), we show in two AD-like mouse models that chronic intermittent increases or reductions of activity have opposite effects on Aβ deposition. Neural activity reduction markedly decreases Aβ aggregation in regions containing axons or dendrites of DREADD-expressing neurons, suggesting the involvement of synaptic and nonsynaptic Aβ release mechanisms. Importantly, activity attenuation is associated with a reduction in axonal dystrophy and synaptic loss around amyloid plaques. Thus, modulation of neural activity could constitute a potential therapeutic strategy for ameliorating amyloid-induced pathology in AD.
SIGNIFICANCE STATEMENT A novel chemogenetic approach to upregulate and downregulate neuronal activity in Alzheimer’s disease (AD) mice was implemented. This led to the first demonstration that chronic intermittent attenuation of neuronal activity in vivo significantly reduces amyloid deposition. The study also demonstrates that modulation of β-amyloid (Aβ) release can occur at both axonal and dendritic fields, suggesting the involvement of synaptic and nonsynaptic Aβ release mechanisms. Activity reductions also led to attenuation of the synaptic pathology associated with amyloid plaques. Therefore, chronic attenuation of neuronal activity could constitute a novel therapeutic approach for AD.
“Attenuation of β-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity” by Peng Yuan (袁鹏) and Jaime Grutzendler in Journal of Neuroscience. Published online January 13 2016 doi:10.1523/JNEUROSCI.2531-15.2016




